
CE MDD | Medical Device
The medical Device Directive is intended to harmonize the legislation on medical devices within the European Union. Legally, in order to sell Medical devices to the European Market, manufacturers must comply with the requirements of the Medical Device Directive. The applicant’s product and quality system must be evaluated, and the manufacturer must affix the CE mark before selling the products.
-
-
-
Applicable Items
The Medical Device Directive is applicable to devices according to the definition of ‘medical device’. A medical device is a machine, device, equipment, material, or other article used alone or in combination that contains software intended for human use as intended by the manufacturer.
Is applicable to diagnosis, prevention, monitoring, treatment or alleviation, diagnosis, monitoring, treatment of disease or alleviation or compensation for injury or disability, investigation, replacement or modification of anatomical or physiological processes, control or concepts. It also refers to something that does not achieve its intended primary action in or on the human body by pharmacological, immunological or metabolic means, but can help its function by such means.
-
-
-
Qualification Assessment Process
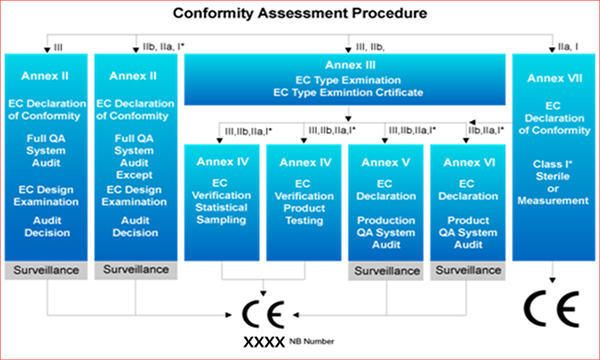 Conformity assessment procedure
✤ Classification
Conformity assessment procedure
✤ ClassificationAnnex IX of the Medical Device Directive(93/42/EEC) provides 18 rules for classifying medical devices. Under these rules, medical devices are classified according to their intended purpose.
- Rule 1 ~ 4 : Non-invasive device
- Rule 5 ~ 8 : Invasive device
- Rule 9 ~ 12 : Active device
- Rule 13 ~ 18 : Special Law
Medical device risk assessment to patients and users are identified and classified according to the above 18 rules. The eligibility assessment process is based on the identified risk values. If the risk value is higher, the eligibility requirements get more stringent.
Class I medical devices follow the evaluation procedures in Annex VII.✤ Procedure of Class I (Including measurement function)- Qualification and technological documentation of competence to meet metrological requirements for each or statistically selected ample in accordance with Annex VII section 3 and Annex IV
- Evaluation and technical documentation of product quality system in accordance with Annex VII section 3 and Annex V which is related to statistical requirements.
- Quality assessment and technical documentation of medical devices in accordance with Annex VII section 3 and Annex VI associated with statistical requirements.
Manufacturer’s option :
✤ Procedure of Class I (including sterilization)- Evaluation and technical documentation of product quality systems in accordance with Annex VII section 3 and annex V associated with sterile conditions.
Manufacturer’s option :
✤ Procedure of Class IIa Medical device- Qualification and technical documentation of each or statistically selected sample according to Annex VII Section 3 and Annex IV Section 8.
- Evaluation and technical documentation of the product quality system in accordance with Annex VII section 3 and Annex V section 6(examined at the manufacturer’s site).
- Evaluation and technical documentation of medical device quality in accordance with Annex VII Section 3 and Annex VI Section 6(examined at on-site audit).
- Evaluation and technical documentation where the overall quality system has been audited at the manufacturer’s site in accordance with Annex VII section3 and Annex II (Development evaluation excluded according to section 4)
Manufacturer’s option :
✤ Procedure of Class IIb Medical Device- Qualification, type examination and technical documentation for each manufactured product in accordance to Annex III Section 3: Samples selected statistically(randomly) according to Annex IV or validated by Section 5.
- Evaluation, type examination and technical documentation of manufacturing quality systemin accordance with Annex III Section 3 and Annex V(examined at onsite audit)
- Technical documentation of evaluation and type examination of medical device in accordance with Annex III Section 3 and Annex VI(examined at manufacturer’s onsite audit)
- Technical documentation of the evaluation of all quality systems that have been audited on site of the manufacturer in accordance with Annex II Section 3.2 and Annex II (excluding development evaluation by Section 4)
Manufacturer’s option :
✤ Procedure of Class III Medical device- Evaluation of all quality systems according to Annex II Section 3.2 and Annex II(examined at the manufacturer’s on site audit) and technical documentation of the description of the development according to Section 4.2.
- Annex III Section 3 and Qualification, Type examination and technical documentation for each manufactured device: Verification shall be made by Section 5 for statistically (randomly) selected samples in accordance with Annex IV Section 6.
- Technical documentation of Evaluation, type examination of the product quality system(examined at the manufacturer’s onsite audit) in accordance with Annex III Section 3 and Annex V Section 3.2
Manufacturer’s option :
-
-
-
End of the MDD
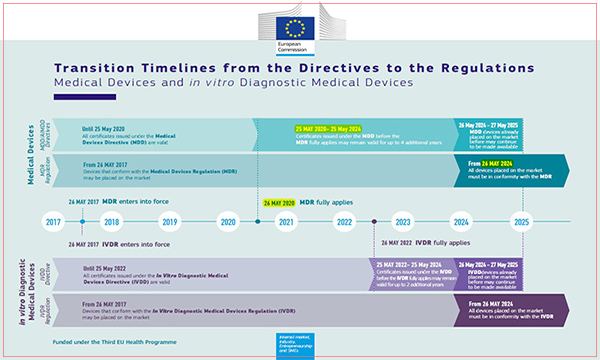
The European Medical Device Directive MDD(93/42/EEC) will be completely replaced by MDR from 26 May 2020. After that, it is not possible to apply for CE certification to MDD, and the new medical device must meet the MDR (2017/745/EU) requirements.
However, if the MDD conformity assessment has been completed and the certificate has been issued before the time of compulsory application, the product can be shipped(exported) to European jurisdiction with the validity of the certificate recognized up to May 27, 2024.
You may check the above materials officially issued by the European Authority.
-
-
-

-
CE / MDD Service
IGC is currently working on a CE/MDD project in cooperation with 3 Notified Body (referred to as NB) organizations. Because of this, you can apply without exception of almost all scopes from Class I to Class III. We will not spare any support to obtain your CE/MDD certification based on the experience accumulated through numerous projects.
-
Related Services from IGC
01System certification (ISO 13485, ISO 15378, ISO 14155)
02Product certification (European CE certification, clinical evaluation, medical device registration [Eurasia, China, USA, Thailand, Taiwan])
03Certification of Auditor Qualifications
04Professional manpower training and education